Introduction To Pharmacology
#1 of 2
September 1999
Go To Section #2 of 2
Last updated 09/06/01 08:53 PM
Keywords & Concepts:
Pharmacodynamics: Read Brody p. 6. What your body
does to a drug, how it augments delivery and absorption, active ingredients and
formulation
Drug receptors: Brody pg. 9. Site of
action, could be membrane protein, cytoplasm or extracellular enzyme or nucleic
acid
Proteins: Brody pg. 9. Usually are
imbedded in cellular subcell. Membrane and facilitate Communication btw the 2
sides of the membrane.
Nucleic acids: Found in the cytoplasm of
cells. ? if this refers to the base of DNA or RNA
Receptor subtypes: For many receptors subtypes
have been identified. They represent variants of the receptor coding sequence ie.
Beta-adrenergic receptors. Some subtypes couple to different signal-generated
molecules and activate separate signaling pathways.
Transmembrane signaling mechanisms:
Lipid soluble drugs (intracellular receptors): The
more lipid soluble a drug the easier it crosses the lipid bilayer membrane. The
receptors for these are inside the cell. Examples are steroids, O2,
nitrous. All can easily cross but still need to interact with some receptor.
Nitrous interacts with or activates guatylate cyclase and this increases cGMP.
Steroids interact with a protein, which interacts to promote a DNA to make a
protein from something else – or gene transcription.
Ligand regulated enzymes (enzyme coupled
receptors): An example is JAK or janus kinase which
phosphorylates STAT- signal transduces and activators of transcription.
Ligand regulated channels (tyrosine kinase coupled
receptors): insulin interacts with a receptor to get tyrosine
kinase activated. Kinases phosphorylate. This sets off a cascade of events
Ligand gated ion channels (ion channel coupled receptors): Ligand
binds or activates the ion channel to open or close like the nicotinic
acetylcholine receptor/channels.
G-protein mediated mechanisms: Regulate
generation of second messenger system. . Binding of an agonist initiates a
sequence of reactions that eventually regulate the activity of membrane-bound
proteins termed effectors.
Intracellular receptor:
Ligand binds to enzyme: ligand binds to a
receptor and activates an enzyme, e.g., tyrosine kinase
Ligand binds to carrier molecules or DNA (transcription
factor): ligand bind to a cytosolic receptor (e.g., chaparone or
heat shock protein) which enters the nucleus and activators (or inhibits)
transcription (gene expression).
Drug-receptor interactions:
Agonist: Brody pg. 15. Ligands that bind
to receptor and activate a process, which ultimately causes an effect. Can be
direct as in open an ion channel or indirect such as a g-protein activates
adenylate cyclase which activates cAMP
Antagonist (blocker): Brody pg. 15.
Binds to same site as agonist but blocks the response. Competitive antagonists
will compete with agonist for same site. The law of mass action will determine
which ligand will win and turn on or off the receptor.
Antagonist (Inhibitor): Brody pg. 15.
See above
Partial agonist: Stimulates a response but
doesn’t do it as well as a full agonist. On a graded-response curve the curve
will stay lower and not reach the peak height of the full agonist.
Ligand: The active ingredient of a drug. It
can be endogenous like epi or norepi or it can be exogenous like isoproterenol.
It is the chemical or molecule involved in the interaction with a specific
receptor.
Binding affinity: A measure of how
"attractive" a ligand is for its receptor. With a higher binding
affinity the ligand would be expected to remain bound to its receptor longer
than a ligand to the same receptor with a lower binding affinity.
Allosteric action: Occurs when a drug binds to
a different site on the extracellular side of the receptor than does the agonist
without producing a signal by itself: however, an enhanced response is produced
when the endogenous ligand binds.
Non-competitive antagonist: Binds to
allosteric site and acts noncompetitively to diminish the signal of the
endogenous agonist.
Competitive antagonist: competes with agonist
for same site and diminishes or blocks the signal.
Reversible binding: Competitive antagonists
are usually weak bonds with low to medium affinity, come on and off with mass
action.
Irreversible binding: Noncompetitive
antagonists that are irreversible have strong covalent bonds, which share
electrons and have high affinity for the binding sites. The alpha adrenergic
receptor phenoxybenzamine doesn’t come off the receptor. It must be taken into
the cell and broken down. (downregulation)
Desensitization: Brody pg. 22. A
decrease in responsiveness of receptor to transmembrane signaling mechanism.
Homologous refers to a specific class of agonists that desensitize its own
receptors and implies modification of receptor itself. Heterologous refers to
several different classes of agents that cause desensitization and implies the
ligand causing the desens may have more widespread effect on signaling system.
Overall desensitization can be caused by a decrease in # of receptors, altered
affinity to binding, phosphorylation of receptor by receptor kinases, or
uncoupling of receptor from 2nd messenger.
Receptor down-regulation: Brody pg. 23.
A decrease in the # of receptors and possible desensitization. It may involve a
decrease in transcription of receptor messenger RNA or an increase in
degradation of mRNA. It can occur after continuous agonist exposure.
Receptor up-regulation: Brody pg. 23.
Receptor supersensitivity occurs after exposure of receptor to antagonist,
inhibition of synthesis, or release of cognate neurotransmitter or hormone. Ie.
Chronic beta blockade will cause an increase in beta-adrenergic receptors on the
heart. The cells will become more sensitive to the agonist.
Drug concentration and responses: magnitude of
response or Emax and is directly proportional to the fraction of
receptors occupied. Emax occurs when all of a given receptor subtype
are occupied by a drug. The magnitude increases as the concentration increases.
Can tell potency and efficacy of drugs compared to each other. There is a linear
response on the curve. Typically at lower concentrations you will see a greater
rate of rise. Quantal response is the frequency of response. # of subjects
responding to a given concentration or dose. Determines the minimum
concentration needed to obtain a response. These curves are better for clinical
comprehension. Look for the median effective curve. This curve shows a black or
white picture – no gray area thus no graded response to measurement. It is the
variability of respondents – not all will respond to the same dose. It will
depend on age, metabolism, fat distribution, protein binding, and gender.
ED50: Brody pg. 31. Effective
dose or concentration where 50% of subjects respond. Doesn’t have to be a
therapeutic or maximal response
KD: the equilibrium constant for
dissociation. Used in linear graph with a dose-response curve and shows that 50%
of binding sites are occupied by drug.
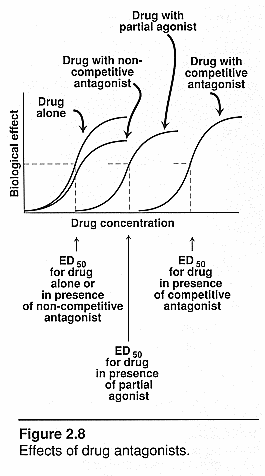
Dose/response curves (drug/receptor
interactions): See notes and this file for actual curves!
Agonists alone: steep log "s" curve
going to Emax
Agonist and competitive antagonist: Both
compete for the same receptor site so the curve will shift to the right and be
less potent. Will need more agonist to overcome antagonist. It can have the same
efficacy and works by the law of mass action.
Agonist and irreversible antagonist: The
initial rise of the s curve will be the same for both but then with the irrev.
Antag the curve will flatten out and go horizontal to the Y-axis. There is a
fixed amt of blocker so it won’t hit every receptor on the tissue. The
receptors that are taken by the noncompetitive blockers will be out of the
picture. The remaining ones will still have all the same properties as those
without any blocker so each receptor acts normal. If all receptors were blocked
then there would be no response. Max response will decrease since there are
fewer receptors available to the agonist.
Agonist and partial agonist: There would be a
backward "s" curve beginning at the top close to the x axis and
dropping down further from the x axis as it reaches the y axis. The partial
agonist will compete with the full agonist so it doesn’t elicit the same
response. It will dampen the effect of the full agonist and will inhibit the
response.
Pharmacokinetics: Brody pg. 6. What your
body does to the drug
Routes of drug administration (advantages and limitations of
each route): Brody pg. 49.
Oral: Absorbed by GI tract and goes to portal
circulation so can be metabolized by the first pass hepatic metabolism through
the liver thus limiting efficacy. Presence of food in the stomach delays gastric
emptying or the drug could be destroyed by acid.
Sublingual: Diffuses into capillary network and
enters systemic circulation thus bypasses 1st pass metabolism
Rectal: 50% of drugs of rectal region bypass
portal circulation
IV- rapid effect and max degree of control but
can induce bacteria, induce hemolysis
IM – aqueous solutions will absorb fast, special
depot preparations will absorb slowly.
SC – slower than IV but can give small doses
of epi to restrict the area of action and decrease removal of the drug from the
area since epi vasoconstricts.
Inhalation, intranasal, intrathecal, topical and
transdermal
Absorption: Brody pg. 35.
Mechanisms of absorption:
Passive
diffusion: Drugs are usually very small and can fit through the
Junctions in the capillary membrane. An example is PCN diffusing passively in
the renal tubules.
Facilitative
diffusion: Ligands can cross membranes using carrier proteins as
in facilitated diffusion.
Active transport: Larger
proteins will require active transport using carrier proteins. The high
molecular wt polypeptides and proteins cannot be administered orally because
there are no mechanisms for their absorption from the GI tract.
Pinocytosis: Large
proteins can enter the cell this way which once the ligand has bound to the cell
membrane, this whole section is invaginated into the cell and becomes engulfed
in the cell.
Lipid Solubility: Drugs that have high lipid
solubility cross membranes better than those with low lipid solubility. Drugs
with high water solubility will have to enter the cell through the watery
channels or other means. Body fluid is aqueous and so hydrophilic substances
will distribute evenly and easily in the blood and can be transported easily and
exchanged through different body compartments.
Degree of ionization: A charged molecule will
attract water (polar substance) and will be stuck to an acid or base when it is
charged. Charged molecules are therefore more water soluble (aqueous). If you
want a drug to be carried in the blood stream which is an aqueous medium then
make it more water soluble. If you want the drug to cross the cell membrane
barrier easier, then make it uncharged or more lipid soluble. Uncharged
substances will cross the membrane barrier. In general an acid in a more acidic
environment is uncharged and in general a base in a more basic environment is
uncharged. Most drugs are weak bases. The passive diffusion of a weak
electrolyte is a function of the pKa of the drug and the pH of the 2
compartments btw which the drug will be moving. In general a weak base with a pH
> pKa will be uncharged and an acid with a pH < pKa
will be uncharged. The further away (as is pH being more acidic) from its pKa
the more uncharged it will be. The more acidic an acid is in relation to its pKa,
the more uncharged it will be. Typically the higher the pKa for a
weak acid the weaker the charge is. The Henderson-Hasselbalch equation plays in
here and you can calculate the actual degree of ionization. See Brody, p 37 for
the equation.
Distribution: This is influenced by blood flow
(Q). The vessel rich groups will get the most blood flow so different organs
will receive different amts of CO. The brain will get the most down to adipose
tissue receiving the least. For consideration of whether a drug gets distributed
is to think of target tissue and ? its blood flow.
Bioavailabilty: The fraction of dose reaching
systemic circulation after administration by any route. Will be affected by drug
binding to plasma proteins.
Factors effecting distributions:
Blood flow: The vessel
rich groups will receive a greater portion of the CO of the body which will
affect certain drugs from reaching certain sites at certain rates of speed.
Capillary permeability: Loose
junctions or fenestrations will allow more drug to diffuse through the cell
membranes with less discrimination. Tight junctions do not allow much to pass as
in the blood-brain barrier.
Chemical nature of drugs: Some
drugs are unstable in stomach pH such as insulin or other proteins. The particle
size, salt form, crystal polymorphism, and presence of excipients will also
affect the bioavailability of the drugs.
Drug binding to tissue and plasma
proteins: When a drug is absorbed in the blood stream and it has
a high affinity for the plasma proteins such as albumin, globulin, alpha1,acid
glycoprotein or lipoproteins or RBC’s, its bioavailability will be decreased
and it becomes inactive. Plasma proteins with drug heavily bound but not free to
pass capillary beds. Only way to get to target tissue is if it’s free unless
the target is the blood. The high affinity keeps the drug in the circulatory
system and decreases its distribution. It also changes its half-life since the
globbed on drugs to the plasma proteins won’t be subject to typical enzymatic
metabolism and not be filtered out in the kidney. Typically weak acids have high
affinity to plasma proteins such as ASA; warfarin and weak bases have a low
affinity such as ethanol. Hepatic disease affects the amt of plasma proteins in
the blood by decreasing them. This will make that binding capacity lower and
will lower this reservoir system in the body so you would need to dose someone
lower.
Biotransformation (Metabolism):
Consequences: activation, maintenance of activity,
inactivation of parent drug: Changes in solubility of the parent compound
will affect biotransformation. If you change it from lipid to aqueous you will
change its solubility. Take a more lipid agent and make it more water soluble.
To keep something excreted, make it more water soluble. By making it more water
soluble it will stay in the urine by globbing on to water molecules and if it is
charged then it won’t cross back across the lipid bilayer. So charged
substances will be excreted. The parent drug is metabolized in the liver usually
and the metabolite can increase or decrease the activity of the drug. L-dopa is
metabolized to dopa, which has an increase in activity that L-dopa.
Benzodiazepines like diazepam and their metabolite have the same activity level.
First pass hepatic metabolism: Oral drugs are absorbed in
the GI tract and go directly through the portal circulation to the liver and are
metabolized there first and then released into the systemic circulation. This is
designed to filter out toxins to the body but in the meantime it can also
decrease the bioavailability of many drugs. NTG is 90% metabolized in the liver
on the 1st pass.
Phase 1 reactions: Brody pg. 40.
Microsomal mixed function oxidase
system: Phase I takes the compound and exposes its functional group by
either globbing something on it or by pulling something off it. This will
increase the water solubility of the compound, which allows it to then by
excreted easier.
Oxidation reactions (cytochrome P450 system dependent and independent):
Reductions and Hydrolysis: Brody pg. 41. The P450 group is a
group of enzymes primarily located in the liver and is responsible for Phase I
biotransformation. The enzymes are inducible or exposable. Drugs can upregulate
or downregulate by increasing the metabolism of all drugs that get metabolized
by that group of cytochromes. CYP3A and CYP3B are very susceptible to up and
downregulation which takes care of about 60% of all clinically relevant drugs.
They are highly inducible. Usually the drugs once metabolized are less toxic;
however, acetaminophen in high doses can be hepatotoxic. The substrates and
saturability if too much of a toxic metabolite and not enough enzymes to drive
detoxification will have an accumulation of the toxic substance. Acetaminophen
goes through Phase I, has a toxic metabolite that goes through Phase II and is
conjugated by glutathione (GSH) and is detoxified. If you run out of GSH then
toxicity will develop. ETOH consumption will enhance this toxicity since it
saturates the enzymes for Phase II and exacerbates this accumulation. Tobacco
smoke and charbroiled food will upregulate CYP1A that takes care of things like
acetaminophen. Glucocorticoids upregulate CYP3A which also handle Phase I
metabolism of acetaminophen, lidocaine and many other anti-arrhythmic drugs.
Thus it will increase the metabolism of them and will increase the amt of toxic
metabolites of acetaminophen. Grapefruit juice inhibits this activity which will
decrease the capability of CYP3A and will decrease the activity for the drugs it
metabolizes thus you could get toxic faster.
Phase 2 reactions:
Conjugation
reactions: This adds something to the compound using glucanuric
acid, amino acids, and glutathione. This coupling of the drug to endogenous
substituent group results will have higher water solubility or other
modification to lead to renal or biliary elimination. This also uses high-energy
phosphate compounds.
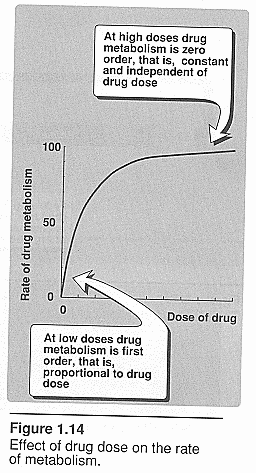
First order kinetics: Brody pg. 55.
Deals with drug clearance. The rate increases directly with an increase in
concentration; the peak concentration is when rates of absorption and
elimination are equal. Rate varies with the 1st power of
concentration.
Zero order kinetics: Brody pg. 55. See graphic above.
Rate is independent of concentration. It can become saturated and rate of
metabolism becomes independent of concentration.
Factors effecting biotransformation: The
following refer to differences in drug pharmacokinetics and pharmacodynamics,
e.g., differences in absorption, distribution, metabolism, etc…
Enzyme
induction: Environment chemicals, air pollutants, cig smoke
stimulate synthesis of higher concentrations of drug metabolizing enzymes.
Genetic
variability:
Age
dependency:
Sex
differences:
Drug
interactions:
First pass
hepatic elimination: Brody pg. 55.
Disease
(e.g., hepatic, renal):
Go To Section #2 of 2
Return To The MNA 2001 Homepage